
By Tien Nguyen, Department of Chemistry
Using a powerful combination of techniques from biophysics to mathematics, researchers have revealed new insights into the mechanism of a liver enzyme that is critical for human health. The enzyme, phenylalanine hydroxylase, turns the essential amino acid phenylalanine – found in eggs, beef and many other foods and as an additive in diet soda —into tyrosine, a precursor for multiple important neurotransmitters.
“We need phenylalanine hydroxylase to control levels of phenylalanine in the blood because too much is toxic to the brain,” said Steve Meisburger, lead author on the study and a post-doctoral researcher in the Ando lab. Genetic mutations in phenylalanine hydroxylase can lead to disorders such as phenylketonuria, an inherited condition that can cause intellectual and behavioral disabilities unless detected at birth and managed through dietary restrictions.
Published earlier this month in the Journal of the American Chemical Society, the article presented detailed structural data on the enzyme’s active state – the shape it adopts when performing its chemical duties – that has eluded scientists for years.
“It’s a floppy enzyme which means it’s dynamic,” said Nozomi Ando, an assistant professor of chemistry at Princeton and corresponding author on the paper. “That also means it doesn’t like to crystallize,” she said. This is problematic for the classic method used to study enzymatic structure, known as x-ray crystallography, which requires solid crystal samples. Efforts to crystallize phenylalanine hydroxylase have just recently met success, but still only captured the enzyme in its inactive state.
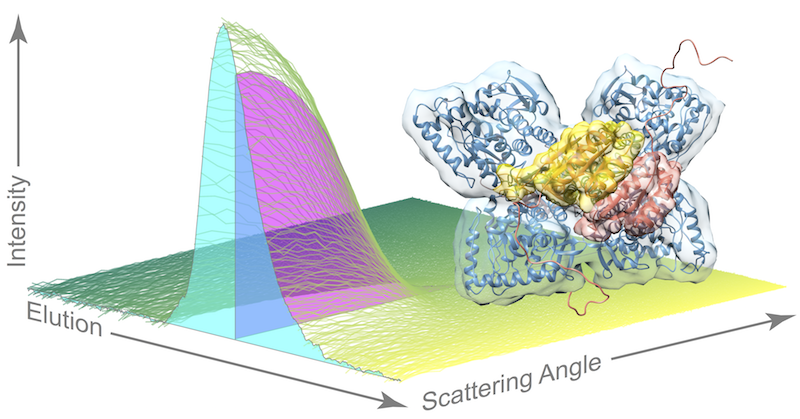
The researchers in the Ando lab were able to bypass the tricky task of growing crystals of the active enzyme by using their expertise in a special technique akin to crystallography, called small angle x-ray scattering (SAXS), which allows scientists to study enzymes in a solution. And because the enzyme is susceptible to aggregation or clumping up in solution, the researchers coupled their scattering method with a purification technique called size exclusion chromatography (SEC), in which different species in a sample flow through a column at different speeds based on their size.

“Pairing SEC with SAXS is an emergent technique. Our contribution is that we saw a clever way to use it,” Ando said. The experiment is highly specialized and relies on powerful x-rays emitted by particles speeding around the circular track at a synchrotron facility. The research team traveled from Princeton to the Cornell High Energy Synchrotron Source in Ithaca, New York, for multiple intensive data-collection sessions. “Any time on the machine that is available, we use it. Not a single photon gets wasted,” Ando said.
As the enzyme solution passes through the purification technique, flowing across the path of the x-ray beam, researchers record snapshots of the x-ray scattering patterns. The resulting dataset is quite complex as the sample also contains phenylalanine, the compound that “turns on” phenylalanine hydroxylase so that researchers can catch the dynamic enzyme in action.
“Current approaches for analyzing this type of dataset are very crude,” Meisburger said. Essentially, these methods assume that each signal – known as an elution peak – represents a single species, when each peak is actually a mixture of species. In this work, the team used an advanced linear algebra method known as evolving factor analysis that allowed them to separate the scattering components. “We can use these linear algebra methods to ‘un-mix’ species that are overlapping,” Meisburger said, “That’s the piece that I think is really exciting.”
By applying their unique approach, the researchers were able to provide evidence for a model of the active structure of phenylalanine hydroxylase that builds upon recent work by their collaborators in Paul Fitzpatrick’s group at UT Health Science Center at San Antonio. In this model, two phenylalanine molecules dock to a pair of sites on the enzyme, bringing a pair of arms together and freeing up the active sites for doing chemistry once more phenylalanine molecules come along.
“I’m very proud that this is our first paper [published since Ando joined the faculty at Princeton]. We wanted it to be very quantitative and heavy on the biochemistry plus heavy on the physical chemistry. I’m really pleased with the way it turned out,” Ando said.
This work was supported by National Health Institutes grants GM100008 and GM098140 and Welch Foundation grant AQ-1245.
Access the paper here:
Meisburger, S. P.; Taylor, A. B.; Khan, C. A.; Zhang, S.; Fitzpatrick, P. F.; Ando, N. “Domain movements upon activation of phenylalanine hydroxylase characterized by crystallography and chromatography-coupled small-angle X-ray scattering.”J. Am. Chem. Soc., 2016, 138 (20), pp 6506–6516.DOI: 10.1021/jacs.6b01563. Published online May 4, 2016.
You must be logged in to post a comment.