
By Catherine Zandonella, Office of the Dean for Research
Genomic sequencing has provided an enormous amount of new information, but researchers haven’t always been able to use that data to understand living systems.
Now a group of researchers has used mathematical analysis to figure out whether two proteins interact with each other, just by looking at their sequences and without having to train their computer model using any known examples. The research, which was published online today in the journal Proceedings of the National Academy of Sciences, is a significant step forward because protein-protein interactions underlie a multitude of biological processes, from how bacteria sense their surroundings to how enzymes turn our food into cellular energy.
“We hadn’t dreamed we’d be able to address this,” said Ned Wingreen, Princeton University‘s Howard A. Prior Professor in the Life Sciences, and a professor of molecular biology and the Lewis-Sigler Institute for Integrative Genomics, and a senior co-author of the study with Lucy Colwell of the University of Cambridge. “We can now figure out which protein families interact with which other protein families, just by looking at their sequences,” he said.
Although researchers have been able to use genomic analysis to obtain the sequences of amino acids that make up proteins, until now there has been no way to use those sequences to accurately predict protein-protein interactions. The main roadblock was that each cell can contain many similar copies of the same protein, called paralogs, and it wasn’t possible to predict which paralog from one protein family would interact with which paralog from another protein family. Instead, scientists have had to conduct extensive laboratory experiments involving sorting through protein paralogs one by one to see which ones stick.
In the current paper, the researchers use a mathematical procedure, or algorithm, to examine the possible interactions among paralogs and identify pairs of proteins that interact. The method was able to correctly predict 93% of the protein-protein paralog pairs that were present in a dataset of more than 20,000 known paired protein sequences, without being first provided any examples of correct pairs.
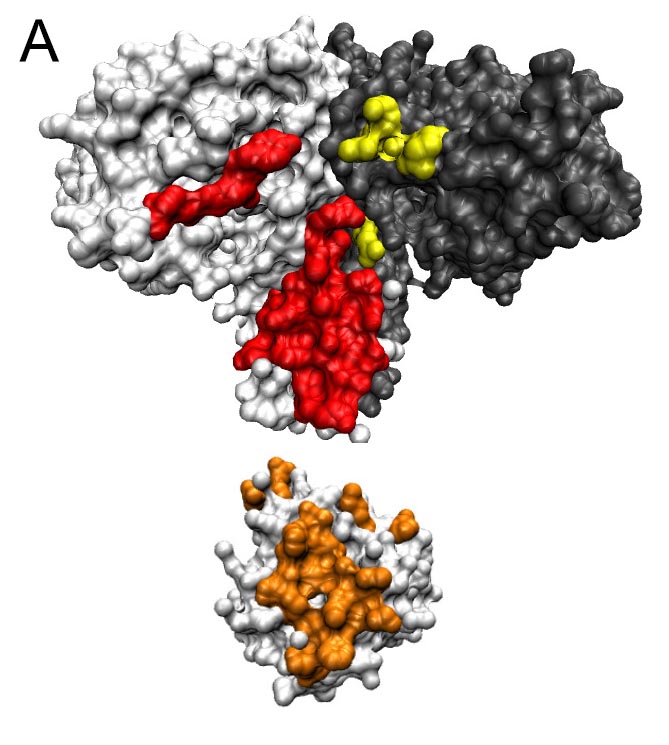
Interactions between proteins happen when two proteins come into physical contact and stick together via weak bonds. They may do this to form part of a larger piece of machinery used in cellular metabolism. Or two proteins might interact to pass a signal from the exterior of the cell to the DNA, to enable a bacterial organism to react to its environment.
When two proteins come together, some amino acids on one chain stick to the amino acids on the other chain. Each site on the chain contains one of 20 possible amino acids, yielding a very large number of possible amino-acid pairings. But not all such pairings are equally probable, because proteins that interact tend to evolve together over time, causing their sequences to be correlated.
The algorithm takes advantage of this correlation. It starts with two protein families, each with multiple paralogs in any given organism. The algorithm then pairs protein paralogs randomly within each organism and asks, do particular pairs of amino acids, one on each of the proteins, occur much more or less frequently than chance? Then using this information it asks, given an amino acid in a particular location on the first protein, which amino acids are especially favored at a particular location on the second protein, a technique known as direct coupling analysis. The algorithm in turn uses this information to calculate the strengths of interactions, or “interaction energies,” for all possible protein paralog pairs, and ranks them. It eliminates the unlikely pairings and then runs again using only the top most likely protein pairs.
The most challenging part of identifying protein-protein pairs arises from the fact that proteins fold and kink into complicated shapes that bring amino acids in proximity to others that are not close by in sequence, and that amino-acids may be correlated with each other via chains of interactions, not just when they are neighbors in 3D. The direct coupling analysis works surprisingly well at finding the true underlying couplings that occur between neighbors.
The work on the algorithm was initiated by Wingreen and Robert Dwyer, who earned his Ph.D. in the Department of Molecular Biology at Princeton in 2014, and was continued by first author Anne-Florence Bitbol, who was a postdoctoral researcher in the Lewis-Sigler Institute for Integrative Genomics and the Department of Physics at Princeton and is now a CNRS researcher at Universite Pierre et Marie Curie – Paris 6. Bitbol was advised by Wingreen and Colwell, an expert in this kind of analysis who joined the collaboration while a member at the Institute for Advanced Study in Princeton, NJ, and is now a lecturer in chemistry at the University of Cambridge.
The researchers thought that the algorithm would only work accurately if it first “learned” what makes a good protein-protein pair by studying ones discovered in experiments. This required that the researchers give the algorithm some known protein pairs, or “gold standards,” against which to compare new sequences. The team used two well-studied families of proteins, histidine kinases and response regulators, which interact as part of a signaling system in bacteria.
But known examples are often scarce, and there are tens of millions of undiscovered protein-protein interactions in cells. So the team decided to see if they could reduce the amount of training they gave the algorithm. They gradually lowered the number of known histidine kinase-response regulator pairs that they fed into the algorithm, and were surprised to find that the algorithm continued to work. Finally, they ran the algorithm without giving it any such training pairs, and it still predicted new pairs with 93 percent accuracy.
“The fact that we didn’t need a gold standard was a big surprise,” Wingreen said.
Upon further exploration, Wingreen and colleagues figured out that their algorithm’s good performance was due to the fact that true protein-protein interactions are relatively rare. There are many pairings that simply don’t work, and the algorithm quickly learned not to include them in future attempts. In other words, there is only a small number of distinctive ways that protein-protein interactions can happen, and a vast number of ways that they cannot happen. Moreover, the few successful pairings were found to repeat with little variation across many organisms. This it turns out, makes it relatively easy for the algorithm to reliably sort interactions from non-interactions.
Wingreen compared this observation – that correct pairs are more similar to one another than incorrect pairs are to each other – to the opening line of Leo Tolstoy’s Anna Karenina, which states, “All happy families are alike; each unhappy family is unhappy in its own way.”
The work was done using protein sequences from bacteria, and the researchers are now extending the technique to other organisms.
The approach has the potential to enhance the systematic study of biology, Wingreen said. “We know that living organisms are based on networks of interacting proteins,” he said. “Finally we can begin to use sequence data to explore these networks.”
The research was supported in part by the National Institutes of Health (Grant R01-GM082938) and the National Science Foundation (Grant PHY–1305525).
The paper, “Inferring interaction partners from protein sequences,” by Anne-Florence Bitbol, Robert S. Dwyerd, Lucy J. Colwell and Ned S. Wingreen, was published in the Early Edition of the journal Proceedings of the National Academy of Sciences on September 23, 2016.
doi: 10.1073/pnas.1606762113
You must be logged in to post a comment.