
By Catherine Zandonella, Office of the Dean for Research
Cells push out tiny feelers to probe their physical surroundings, but how much can these tiny sensors really discover? A new study led by Princeton University researchers and colleagues finds that the typical cell’s environment is highly varied in the stiffness or flexibility of the surrounding tissue, and that to gain a meaningful amount of information about its surroundings, the cell must move around and change shape. The finding aids the understanding of how cells respond to mechanical cues and may help explain what happens when migrating tumor cells colonize a new organ or when immune cells participate in wound healing.
“Our study looks at how cells literally feel their way through an environment, such as muscle or bone,” said Ned Wingreen, Princeton’s Howard A. Prior Professor in the Life Sciences and professor of molecular biology and the Lewis-Sigler Institute for Integrative Genomics. “These tissues are highly disordered on the cellular scale, and the cell can only make measurements in the immediate area around it,” he said. “We wanted to model this process.” The study was published online on July 18 in the journal Nature Communications.
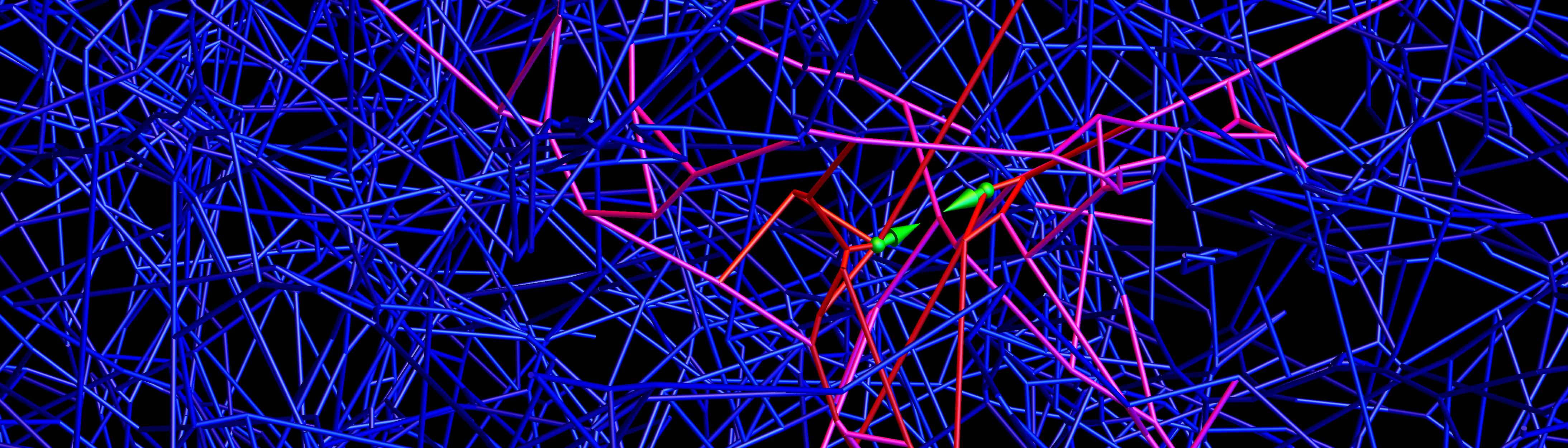
The organs and tissues of the body are enmeshed in a fiber-rich structure known as the extracellular matrix, which provides a scaffold for the cells to live, move and differentiate to carry out specific functions. Cells interact with this matrix by extending sticky proteins out from the cell surface to pull on nearby fibers. Previous work, mostly employing artificial flat surfaces, has shown that cells can use this tactile feedback to determine the elasticity or stiffness in a process called mechanosensing. But because the fibers of the natural matrix are all interconnected in a jumbled, three-dimensional network, it was not clear how much useful information the cell could glean from feeling its immediate surroundings.
To find out, the researchers built a computer simulation that mimicked a typical cell in a matrix made of collagen protein, which is found in skin, bones, muscles and connective tissue. The team also modeled a cell in a network of fibrin, the strong, stringy protein that makes up blood clots. To accurately capture the composition of these networks, the researchers worked with Chase Broedersz, a former Princeton Lewis-Sigler Fellow who is now professor of physics at Ludwig-Maximilians-University of Munich, and his colleagues Louise Jawerth and Stefan Münster to first create physical models of the matrices, using approaches originally developed in the group of collaborator David Weitz, a systems biologist at Harvard University. Princeton graduate student Farzan Beroz then used those models to recreate virtual versions of the collagen and fibrin networks in computer models.
With these virtual networks, Beroz, Broedersz and Wingreen could then ask the question: can cells glean useful information about the elasticity or stiffness of their environment by feeling their surroundings? If the answer is yes, then the finding would shed light on how cells can change in response to those surroundings. For example, the work might help explain how cancer cells are able to detect that they’ve arrived at an organ that has the right type of scaffold to support tumor growth, or how cells that arrive at a wound know to start secreting proteins to promote healing.
Using mathematics, the researchers calculated how the networks would deform when nearby fibers are pulled on by cells. They found that both the collagen and fibrin networks contained configurations of fibers with remarkably broad ranges of collective stiffness, from rather bendable to very rigid, and that these regions could be immediately next to each other. As a result, the cell could have two nearby probes whereby one detects hardness and the other detects softness, making it difficult for a cell to learn by mechanosensing what type of tissue it inhabits. “We were surprised to find that the cell’s environment can vary quite a lot even across a small distance,” Wingreen said.
The researchers concluded that to obtain an accurate assessment of its environment, a cell must move around and also change shape, for example elongating to cover a different area of the matrix. “What we found in our simulation conforms to what experimentalists have found,” Wingreen said, “and reveals new, ‘intelligent’ strategies that cells could employ to feel their way through tissue environments.”
The study was supported in part by the National Science Foundation (grants DMR-1310266, DMR-1420570, PHY-1305525 and PHY-1066293) the German Excellence Initiative, and the Deutsche Forschungsgemeinschaft.
The study, “Physical limits to biomechanical sensing in disordered fiber networks,” by Farzan Beroz, Louise Jawerth, Stefan Münster, David Weitz, Chase Broedersz, and Ned Wingreen, was published in the journal Nature Communications on July 18, 2017. DOI 10.1038/NCOMMS16096.
You must be logged in to post a comment.