
Article provided by the Department of Molecular Biology
Hepatitis delta virus (HDV) causes the most aggressive form of viral hepatitis in humans, putting at least 20 million people worldwide at risk of developing liver fibrosis, cirrhosis, and liver cancer. Efforts to develop effective treatments against HDV have been hampered by the fact that laboratory mice are not susceptible to the virus. But, in a study published June 27, 2018, in the journal Science Translational Medicine, Alexander Ploss, assistant professor of molecular biology at Princeton University and colleagues describe a genetically engineered mouse that can be persistently infected with HDV.
HDV is a small, RNA-based “satellite” virus that produces just a single protein of its own and therefore requires additional proteins provided by another liver virus, hepatitis B virus (HBV). HDV can infect patients already carrying HBV, or both viruses can infect patients simultaneously. Though infections can be prevented with an anti-HBV vaccine, there are no antiviral therapies available to cure existing HDV infections.
HDV and HBV infect the liver by binding to a protein called NTCP that is present on the surface of liver cells. But the viruses only recognize the version of NTCP present in humans and a few other primates, and therefore can’t infect mice or other small mammals that produce their own versions of NTCP. This has made it difficult to study HBV and HDV infections in the laboratory. Researchers have tried transplanting human liver cells into immunocompromised mice before infecting them with virus, but this approach has produced inconsistent results and is both expensive and time-consuming.
Ploss and colleagues, led by graduate student Benjamin Winer, took a different approach. They generated mice that express the human NTCP protein in their liver cells, allowing these cells to be infected by HBV and HDV.
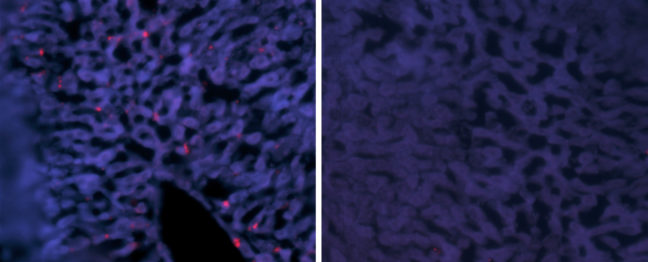
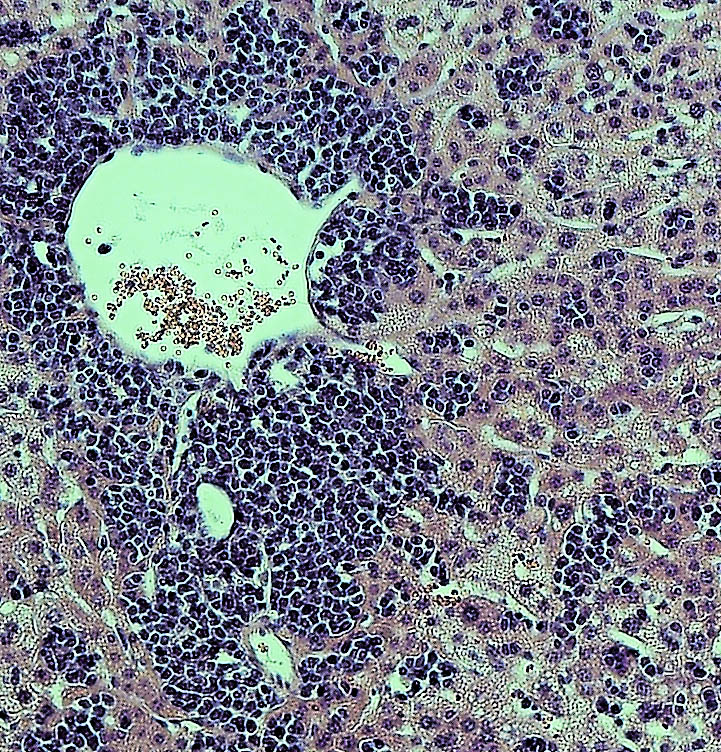
In these mice, HBV failed to replicate after entering mouse liver cells but HDV was able to establish persistent infection when provided with the HBV proteins it needs to propagate. For example, mice genetically engineered to produce both human NTCP and the entire HBV genome could be infected with HDV for up to 14 days. “To our knowledge, this is the first time the entire HDV life cycle has been recapitulated in a mouse model with inheritable susceptibility to HDV,” Ploss said.
The mice were able to rid themselves of HDV before they developed any liver damage, apparently by mounting an immune response involving antiviral interferon proteins and various white blood cell types, including Natural Killer (NK) cells and T cells. Accordingly, mice expressing human NTCP and the HBV genome, but lacking functional B, T, and NK cells could be infected with HDV for two months or more.
These immunocompromised animals allowed Ploss and colleagues to test the effectiveness of two drugs that are currently being developed as treatments for HDV infection. Both drugs—either alone or in combination—suppressed the levels of HDV in immunocompromised mice after viral infection. But the drugs were not able to completely clear the mice of HDV; viral levels rose again within weeks of stopping treatment.
“This is largely in line with recently reported data from clinical trials, showing the utility of our model for preclinical antiviral drug testing,” Winer said.
“Our model is amenable to genetic manipulations, robust, and can be adopted as a method to rapidly screen for potential treatments,” Ploss added.
Timothy M. Block, president of the Hepatitis B Foundation and its Baruch S. Blumberg Institute who was not involved in the study, said “These systems should be able to provide practical, and presumably economical tools. Their work is urgently needed, and a desperate community welcomes it. I emphasize that it is often the new methods in science that revolutionize a field such as drug discovery, almost as much as the new drugs themselves.”
The research team included collaborators from Princeton University; Weill Medical College of Cornell University; The Jackson Laboratory; University Medical Center Hamburg-Eppendorf, Hamburg; New York University Medical Center; and North Carolina State University College of Veterinary Medicine.
This study was supported by grants from the National Institutes of Health (R01 AI079031, R01 AI107301, R21AI117213 to Alexander Ploss), a Research Scholar Award from the American Cancer Society (RSG-15-048-01-MPC to Alexander Ploss), a Burroughs Wellcome Fund Award for Investigators in Pathogenesis (to Alexander Ploss) and a Graduate fellowship from the Health Grand Challenge from the Global Health Fund of Princeton University (to Benjamin Y. Winer). The NYU Experimental Pathology Immunohistochemistry Core Laboratory is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant; NIH/NCI P30CA016087 and the National Institutes of Health S10 Grants NIH/ORIP S10OD01058 and S10OD018338. Benjamin Y. Winer is a recipient of F31 NIH/NRSA Ruth L. Kirschstein Predoctoral awarded from the NIAID. Julie Sellau and Elham Shirvani-Dastgerdi are both recipients of postdoctoral fellowships from the German Research Foundation. Michael V. Wiles was funded by The Jackson Laboratory.
Benjamin Y. Winer, Elham Shirvani-Dastgerdi, Yaron Bram, Julie Sellau, Benjamin E. Low, Heath Johnson, Tiffany Huang, Gabriela Hrebikova, Brigitte Heller, Yael Sharon, Katja Giersch, Sherif Gerges, Kathleen Seneca, Mihai-Alexandru Pais, Angela S. Frankel, Luis Chiriboga, John Cullen, Ronald G. Nahass, Marc Lutgehetmann, Jared Toettcher, Michael V. Wiles, Robert E. Schwartz, and Alexander Ploss. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for persistent hepatitis delta virus infection. Science Translational Medicine. 2018. DOI: 10.1126/scitranslmed.aap9328


You must be logged in to post a comment.