
By the Department of Molecular Biology
Princeton University researchers have uncovered a critical role for a new immune signaling pathway in controlling infection by the flavivirus Yellow Fever Virus (YFV). The paper describing this discovery was published today in the journal mBio.
Infection with YFV causes a devastating illness with a mortality rate of up to 50%. Like other members of its viral family—which includes West Nile Virus, Dengue Virus and Zika Virus—YFV is transmitted to humans by mosquitos that are expanding into new areas across the globe, exposing more people to these dangerous viruses. Fortunately, there is an effective vaccine for YFV: a live-attenuated strain of the virus, called YFV-17D, which differs by only a few amino acids from the virulent viral strain YFV-Asibi, but nonetheless provokes a potent and durable protective immune response in humans.
“An improved understanding of the complex mechanisms regulating YFV-17D attenuation will provide insights into key viral-host interactions that regulate host immune responses and infection outcomes, [and] open novel avenues for the development of innovative vaccine strategies,” said Alexander Ploss, assistant professor at Princeton’s Department of Molecular Biology, who led the study with first author Florian Douam, a postdoctoral research associate. However, research efforts have been hampered due to the fact that mice, which are used in the study of viral infections, are resistant to YFV infection. Nonetheless, recent mouse experiments have pointed to an important role for cytokines called interferons (IFN) in controlling the virus.
Mice, like humans, possess three types of interferons, molecules produced by the immune system during infection: type I interferons, which signal through the widely distributed IFN-α/β receptor; type II interferons that act on IFN-γ receptors present in most tissues; and type III interferons, which activate signaling by IFN-λ receptors found on epithelial cells. Mice lacking type I receptors die after infection by YFV-Asibi, but survive YFV-17D infection despite extensive viral replication at early stage of infection.Type II IFN signaling has also been shown to be important for clearing up late stage YFV-Asibi and YFV-17D infection when type I IFN signaling is defective. By contrast, the contribution of type III IFN signaling to control of YFV infection was unknown.
To address this question, Douam and colleagues studied YFV-17D infection in mice lacking the type III receptor. Initial experiments showed that these mice were able to control viral replication and rapidly cleared YFV-17D, indicating that type III signaling alone wasn’t necessary for resistance to YFV-17D. However, mice lacking both type I and type III receptors succumbed after YFV-17D infection, suggesting type III signaling does contribute to the antiviral immune response.
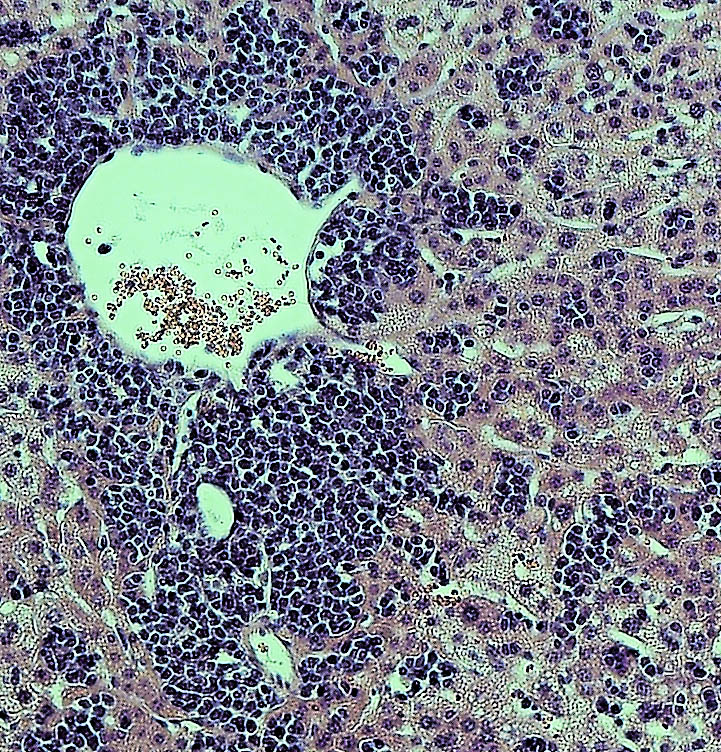
To find out more, the authors examined YFV-17D levels in various tissues. Early in the infection, the virus was present in every tissue of each mouse model examined. However, although viral loads were low in wild-type mice and type III receptor-deficient mice, they were much higher in type I and type I/III receptor-deficient mice. Surprisingly, the viral loads in brains of type I/III receptor-deficient mice increased over time in comparison to type I receptor-deficient mice, showing that loss of type III IFN signaling enhances the susceptibility of type I-receptor deficient animals to brain infection. This is significant because the presence of viruses in the brain can cause brain damage such as spongiosis or encephalitis. The low level of YFV-17FD brain invasion in wild-type mice caused mild spongiosis, whereas type I/III receptor-deficient mice had severe spongiosis—potentially explaining YFV-17D lethality in those animals. However, this raised the question of why YFV-17 was present at such high levels in the animals’ brains.

Another study recently showed that type III IFN signaling affects the epithelial cells that make up the blood brain barrier (BBB), and modulates BBB integrity during infection by another flavivirus, West Nile Virus. Consistent with this, Ploss’s group observed that the BBB of type I/III receptor-deficient mice was especially leaky to a blue dye. But this wasn’t the only way that loss of type III IFN signaling impaired the body’s response to YFV; the researchers also found evidence that type III receptor deficiency provokes strong imbalances in several different kinds of immune cells during YFV-17D infection. In particular, type I/III receptor-deficient mice were defective in the activation of T cells, critical immune cells that control YFV-17D infection.
“We uncovered a critical role of type III IFN-mediated signaling in preserving the integrity of the blood brain barrier and preventing viral brain invasion,” Ploss said. More work is needed to explore how type III IFN signaling affects YFV infection in primates, but this study already provides important new insights about a poorly understood immune signaling pathway.
The study was supported by grants from National Institutes of Health (R01 AI107301, R21AI117213 to Alexander Ploss and R01 AI104669 to Sergei Kotenko). Additional funding included a Research Scholar Award from the American Cancer Society (RSG-15-048-01-MPC), the Princeton Environmental Institute‘s Grand Health Challenge program from Princeton University, and an Investigator in Pathogenesis Award by the Burroughs Wellcome Fund (all to Alexander Ploss).
The study, “Type III Interferon-mediated signaling is critical for controlling live attenuated Yellow Fever Virus infection in vivo.,” by Florian Douam, Yentil E. Soto-Albrecht, Gabriela Hrebikova, Evita Sadimin, Christian Davidson, Sergei V. Kotenko and Alexander Ploss was published in the journal mBio. (2017). doi:10.1128/mBio.00819-17
You must be logged in to post a comment.