
By the Department of Molecular Biology
Whether growing in a puddle of dirty water or inside the human body, large groups of bacteria must coordinate their behavior to perform essential tasks that they would not be able to carry out individually. Bacteria achieve this coordination through a process called quorum sensing in which the microorganisms produce and secrete small molecules called autoinducers that can be detected by neighboring bacterial cells. Only when a large number of bacteria are present can the levels of secreted autoinducer build up to the point where the community can detect them and respond as a coordinated group.
In a paper published last month in PLoS Pathogens, a team of researchers led by postdoctoral research associate Sampriti Mukherjee and professor Bonnie Bassler of the Department of Molecular Biology at Princeton University revealed the existence of a new quorum-sensing molecule that increases the virulence of the pathogenic bacterium Pseudomonas aeruginosa. The finding could help researchers develop new antimicrobial drugs to treat the serious infections caused by this bacterium.
P. aeruginosa is an incredibly adaptable organism that can grow in environments ranging from soil and freshwater to the tissues of plants and animals. It thrives on the surfaces of medical equipment and is therefore a major cause of hospital-acquired infections, causing life-threatening conditions such as pneumonia and sepsis in vulnerable patients. The bacterium has become resistant to commonly used antibiotics, making the development of new antimicrobials a priority for both the Centers for Disease Control and Prevention and the World Health Organization.
Quorum sensing is crucial for P. aeruginosa‘s adaptability. The process regulates the development of biofilms, the three-dimensional structures formed by large bacterial communities that promote their ability to establish and maintain infections. “P. aeruginosa strains harboring mutations in the quorum-sensing machinery are attenuated for virulence, and thus, interfering with quorum sensing holds promise for the development of novel anti-microbial therapies,” said Bassler, the Squibb Professor in Molecular Biology at Princeton University and a Howard Hughes Medical Institute Investigator.
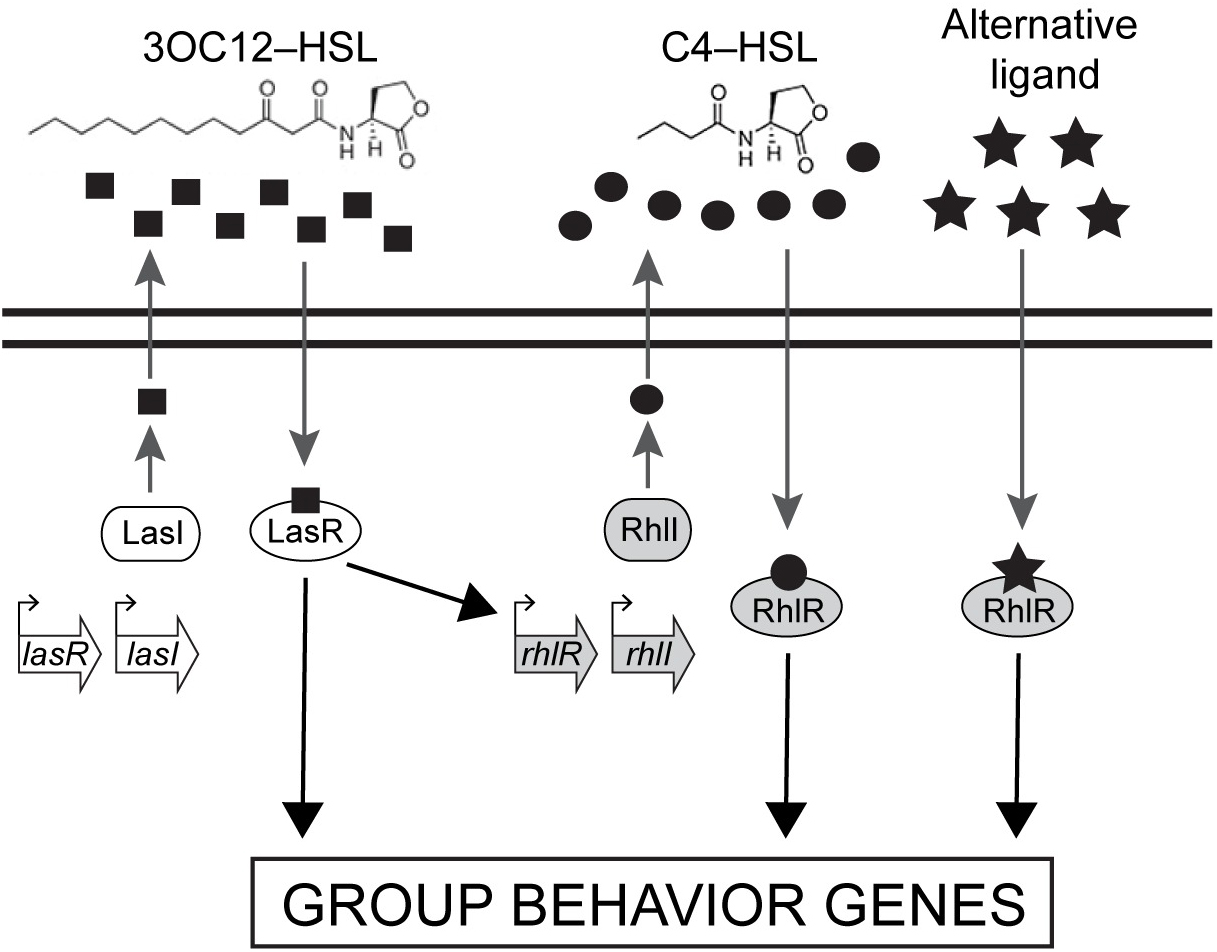
P. aeruginosa possesses similar quorum-sensing machinery to other species of bacteria. For example, it produces an enzyme called RhlI that synthesizes an autoinducer molecule known as N-butanoyl-L-homoserine lactone, or C4-HSL. This molecule can then bind and activate a protein called RhlR that regulates the expression of multiple genes that P. aeruginosa needs to form a biofilm and/or infect a host.
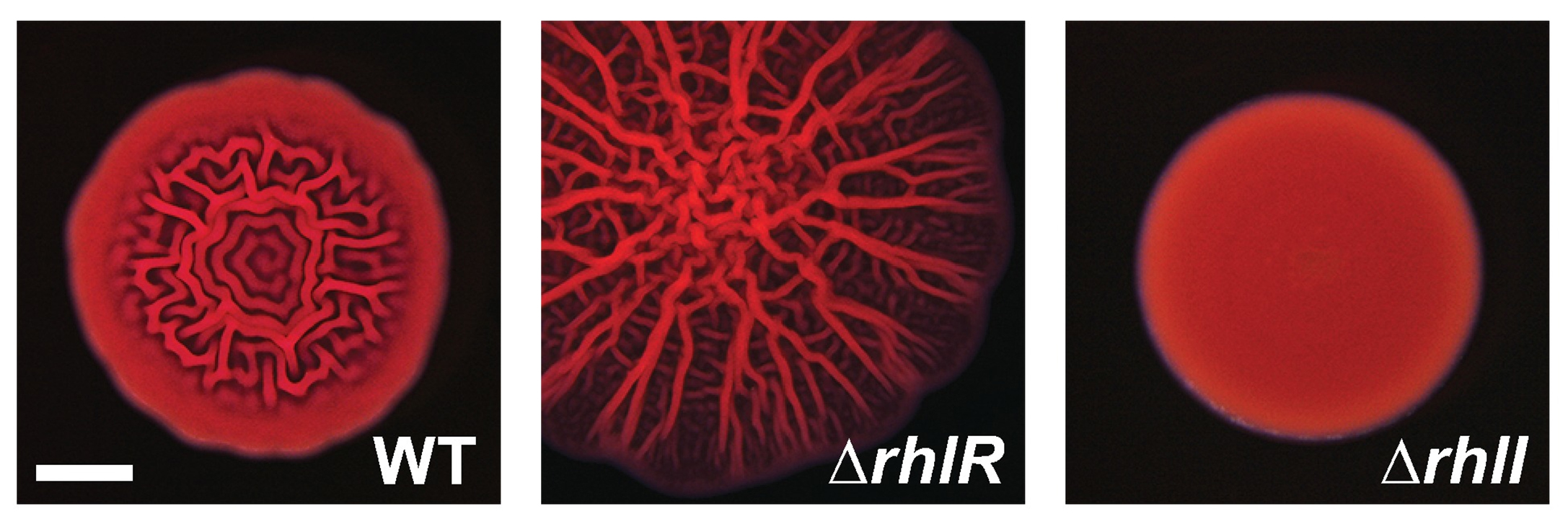
In theory, removing RhlI or RhlR should have the same effect on P. aeruginosa cells, since the latter protein shouldn’t be able to work without the autoinducer produced by the former. But the researchers, led by postdoctoral fellow Sampriti Mukherjee, noticed that bacterial colonies lacking RhlI formed unusually smooth biofilms, whereas strains lacking RhlR formed biofilms that were much more wrinkled than normal.
The researchers went on to show that in biofilms, many genes only depended on RhlR, not on RhlI. “That suggested that RhlR can be activated by an alternative molecule, in addition to C4-HSL,” Bassler said.
The researchers found that bacteria lacking RhlI, which are therefore unable to synthesize the C4-HSL autoinducer, still secrete a molecule capable of activating RhlR. Bassler and colleagues don’t yet know what this molecule is, but it seems to be quite different from C4-HSL. “We are currently working to purify and identify this molecule,” Bassler said.
Crucially, the activation of RhlR by this unknown molecule may be important for P. aeruginosa‘s ability to infect animals. Mukherjee and the team found that bacteria lacking RhlI were just as effective as wild-type bacteria in infecting both roundworms and mice. But bacteria lacking RhlR were much less virulent and far less able to grow inside these animals. “Targeting RhlR with small-molecule inhibitors could provide an exciting path forward for the development of novel antimicrobial drugs,” Bassler said.
The ability of RhlR to be activated by distinct molecules might also help explain P. aeruginosa‘s adaptability. Bassler and colleagues speculate that different environments could stimulate discrete levels of production of the different autoinducer molecules, each of which could activate RhlR, or a related protein called LasR, to induce expression of the specific genes the bacteria need to thrive in that particular location.
The work was supported by the Howard Hughes Medical Institute, the National Institutes of Health (grant 2R37GM065859), the National Science Foundation (grant MCB-0948112), and a Life Science Research Foundation Postdoctoral Fellowship through the Gordon and Betty Moore Foundation (grant GBMF2550.06).
In addition to Mukherjee and Bassler, the team consisted of postdoctoral fellow Dina Moustafa and professor Joanna Goldberg in the Department of Pediatrics at Emory University School of Medicine, and Chari Smith, a research consultant at Princeton University.
The study, “The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer,” by Sampriti Mukherjee, Dina Moustafa, Chari D. Smith, Joanna B. Goldberg, and Bonnie L. Bassler, was published in the journal PLoS Pathogens on July 17, 2017. DOI:10.1371/journal.ppat.1006504.


You must be logged in to post a comment.