

By Tien Nguyen, Department of Chemistry
Researchers at Princeton and Harvard Universities have developed a way to produce the tools for figuring out gene function faster and cheaper than current methods, according to new research in the journal Nature Communications.
The function of sizable chunks of many organisms’ genomes is a mystery, and figuring out how to fill these information gaps is one of the central questions in genetics research, said study author Buz Barstow, a Burroughs-Wellcome Fund Research Fellow in Princeton’s Department of Chemistry. “We have no idea what a large fraction of genes do,” he said.
One of the best strategies that scientists have to determine what a particular gene does is to remove it from the genome, and then evaluate what the organism can no longer do. The end result, known as a whole-genome knockout collection, provides full sets of genomic copies, or mutants, in which single genes have been deleted or “knocked out.” Researchers then test the entire knockout collection against a specific chemical reaction. If a mutant organism fails to perform the reaction that means it must be missing the particular gene responsible for that task.
It can take several years and millions of dollars to build a whole-genome knockout collection through targeted gene deletion. Because it’s so costly, whole-genome knockout collections only exist for a handful of organisms such as yeast and the bacterium Escherichia coli. Yet, these collections have proven to be incredibly useful as thousands of studies have been conducted on the yeast gene-deletion collection since its release.
The Princeton and Harvard researchers are the first to create a collection quickly and affordably, doing so in less than a month for several thousand dollars. Their strategy, called “Knockout Sudoku,” relies on a combination of randomized gene deletion and a powerful reconstruction algorithm. Though other research groups have attempted this randomized approach, none have come close to matching the speed and cost of Knockout Sudoku.
“We sort of see it as democratizing these powerful tools of genetics,” said Michael Baym, a co-author on the work and a Harvard Medical School postdoctoral researcher. “Hopefully it will allow the exploration of genetics outside of model organisms,” he said.
Their approach began with steep pizza bills and a technique called transposon mutagenesis that ‘knocks out’ genes by randomly inserting a single disruptive DNA sequence into the genome. This technique is applied to large colonies of microbes to ensure the likelihood that every single gene is disrupted. For example, the team started with a colony of about 40,000 microbes for the bacterium Shewanella oneidensis, which has approximately 3,600 genes in its genome.
Barstow recruited undergraduates and graduate students to manually transfer 40,000 mutants out of laboratory Petri dishes into separate wells using toothpicks. He offered pizza as an incentive, but after a full day of labor, they only managed to move a couple thousand mutants. “I thought to myself, ‘Wait a second, this pizza is going to ruin me,’” Barstow said.
Instead, they decided to rent a colony-picking robot. In just two days, the robot was able to transfer each mutant microbe to individual homes in 96-well plates, 417 plates in total.
But the true challenge and opportunity for innovation was in identifying and cataloging the mutants that could comprise a whole-genome knockout collection in a fast and practical way.
DNA amplification and sequencing is a straightforward way to identify each mutant, but doing it individually quickly gets very expensive and time-consuming. So the researchers’ proposed a scheme in which mutants could be combined into groups that would only require 61 amplification reactions and a single sequencing run.
But still, after sequencing each of the pools, the researchers had an incredible amount of data. They knew the identities of all the mutants, but now they had to figure exactly where each mutant came from in the grid of plates. This is where the Sudoku aspect of the method came in. The researchers built an algorithm that could deduce the location of individual mutants through its repeated appearance in various row, column, plate-row and plate-column pools.
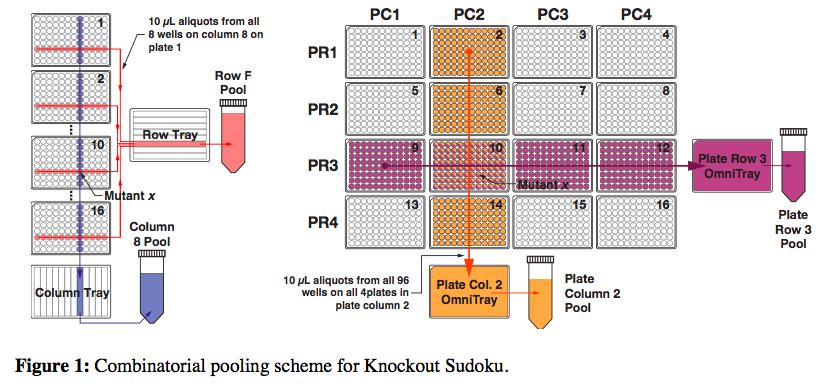
But there’s a problem. Because the initial gene-disruption process is random, it’s possible that the same mutant is formed more than once, which means that playing Sudoku wouldn’t be simple. To find a solution for this issue, Barstow recalled watching the movie, “The Imitation Game,” about Alan Turing’s work on the enigma code, for inspiration.
“I felt like the problem in some ways was very similar to code breaking,” he said. There are simple codes that substitute one letter for another that can be easily solved by looking at the frequency of the letter, Barstow said. “For instance, in English the letter A is used 8.2 percent of the time. So, if you find that the letter X appears in the message about 8.2 percent of the time, you can tell this is supposed to be decoded as an A. This is a very simple example of Bayesian inference.”
With that same logic, Barstow and colleagues developed a statistical picture of what a real location assignment should look like based on a mutant that only appeared once and used that to rate the likelihood of possible locations being real.
“One of the things I really like about this technique is that it’s a prime example of designing a technique with the mathematics in mind at the outset which lets you do much more powerful things than you could do otherwise,” Baym said. “Because it was designed with the mathematics built in, it allows us to get much, much more data out of much less experiments,” he said.
Using their expedient strategy, the researchers created a collection for microbe Shewanella oneidensis. These microbes are especially good at transferring electrons and understanding their powers could prove highly valuable for developing sustainable energy sources, such as artificial photosynthesis, and for environmental remediation in the neutralization of radioactive waste.
Using the resultant collection, the team was able to recapitulate 15 years of research, Barstow said, bolstering their confidence in their method. In an early validation test, they noticed a startlingly poor accuracy rate. After finding no fault with the math, they looked at the original plates to realize that one of the researchers had grabbed the wrong sample. “The least reliable part of this is the human,” Barstow said.
The work was supported by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund and Princeton University startup funds and Fred Fox Class of 1939 funds.
Read the full article here:
Baym, M.; Shaker, L.; Anzai, I. A.; Adesina, O.; Barstow, B. “Rapid construction of a whole-genome transposon insertion collection for Shewanella oneidensis by Knockout Sudoku.” Nature Comm. Available online on Nov. 10, 2016.

You must be logged in to post a comment.